Fasリガンドによるアポトーシスと炎症の分子機構とその意義
(Molecular
Mechanismsand Biological Roles of Apoptosis and
Inflammation Induced by Fas Ligand)
須田貴司
(蛋白質核酸酵素 (1999) Vol.44 p1478-1486)
要約:
Fasリガンドはアポトーシスを誘導するサイトカイン(デス因子)である。近年、デス因子が次々と発見されているが、FasリガンドはTNF(腫瘍壊死因子)とともにデス因子のプロトタイプとして最も盛んに研究されている。様々なアポトーシス誘導系の中で、細胞外のシグナルからカスペースの活性化までの経路が明らかになっているのは、デス因子によるアポトーシス誘導系のみである。また、Fasリガンドは細胞傷害性T細胞の主要なエフェクター分子の一つとして働いている他、免疫系の制御にも重要な役割を果たしていることが明らかになっている。
はじめに
近年アポトーシスという言葉が医学、生物学の領域で盛んに聞かれるようになった。アポトーシスという現象が注目される理由は、それが個体全体の利益のために個々の細胞が自殺するという生命の根源に関わる細胞死だからである。個体発生の過程で機能的な形を刻むために不要な細胞は除かれる。例えば5本の指が出来るのは、指と指の間の細胞がアポトーシスを起こして無くなるからである。また、放射線、熱、薬剤などの物理科学的ストレスにより傷害された細胞やウイルスが感染した細胞等は、そのまま崩壊していけば細胞内から洩れ出したプロテアーゼなどにより周囲の正常な細胞を傷害したり、ウイルス感染を拡大させることになる。このようなとき細胞は内在する自爆の生化学装置を活性化し、染色体を切り刻み、細胞質や核を細切れにして、貪食細胞などに食べられやすくする。これがアポトーシスである。個体発生もホメオスタシスも細胞の増殖、分化、死のバランスの上に成り立っている(図1)。従って、アポトーシスの抑制も過剰も形態形成の異常をきたす。また、アポトーシスの抑制は癌や自己免疫疾患等の原因に、過多はエイズ、劇症肝炎、アルツハイマー病などの神経変性疾患など様々な疾患の原因になると考えられている。アポトーシスは通常炎症を起こしにくい細胞死とされているが、炎症細胞にアポトーシスが起こった場合にはさらに炎症を増強する機構が存在するようである。
|
|
図1 細胞増殖、分化、死のバランス個体発生もホメオスタシスも細胞の増殖、分化、死のバランスの上に成り立っている。細胞の死もこのバランスを保つうえで重要な要素であり、細胞の機能の一つと言える。デス因子の存在は細胞死の機能的重要性を裏付けるものである。細胞死のバランスが崩れることは様々な疾患の原因になる。 |

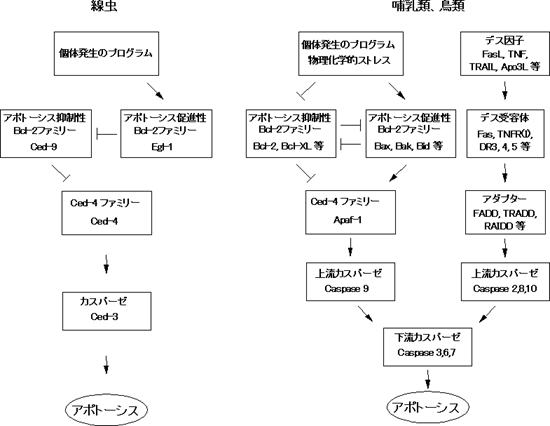
アポトーシスの分子機構は、先ず線虫の遺伝学的解析からその大筋が明らかにされた。Horvitzらは発生過程でのアポトーシスに異常を示す線虫の突然変異を次々に単離し、アポトーシスの決定(ces-1, ces-2, egl-1)、アポトーシスの実行(ced-3, ced-4)、アポトーシスの抑制(ced-9)、死んだ細胞の取込み(ced-1, ced-2)に関与する遺伝子の存在を明らかにした。この内egl-1, ced-3, ced-4, ced-9には構造的にも機能的にも対応するホモログが哺乳類にも存在することが判明し、アポトーシスの分子装置は線虫からヒトまで極めてよく保存されていることが明らかになった ( 図2)。一方、哺乳類と鳥類には、線虫には存在しない細胞死誘導サイトカイン(デス因子)とその受容体を介したアポトーシス誘導機構が存在する。ここでは、デス因子のプロトタイプとも言えるFasリガンドのシグナル伝達機構と生理的・病理的役割についてこれまでの知見を解説する。
|
図2 線虫と高等脊椎動物のアポトーシスシグナル伝達機構 高等脊椎動物は線虫から保存されているBcl-2を介するアポトーシス誘導機構以外にデス因子、デス受容体を介するアポトーシス伝達機構を発達させた。アポトーシス促進性、抑制性Bcl-2ファミリーはそれぞれミトコンドリアからのチトクロムcの放出を促進あるいは抑制すると考えられている。チトクロムcはApaf-1 (apoptotic protease activating
factor-1)に結合することにより、Apaf-1とカスパーゼ9の結合を促進し、その結果カスパーゼ9が活性化する。デス因子シグナルの詳細は本文及び図3参照。 |
I
. Fas-Fasリガンド系のアポトーシス誘導機構
1. Fas、Fasリガンドの構造
FasリガンドはTNF (tumor necrosis factor) に類似の40kDのII型膜貫通蛋白質である(図3)(1, 2)。Fasリガンドの細胞質領域は約80アミノ酸からなり、リガンドとしては長い細胞質領域を持っている。この領域には極めてプロリンに富んだユニークな配列がヒト、マウス、ラットを通じてよく保存されている。細胞質領域を削ったFasリガンド遺伝子を導入すると細胞表面のFasリガンド発現量が著しく増強する。これはこの細胞質領域がFasリガンドを細胞内顆粒にとどめる作用を持っているためであることが、最近明らかにされた。この細胞内Fasリガンドはプールとして働き、細胞外からの刺激に応じて速やかに細胞表面に移動する。細胞外領域は179アミノ酸からなり、カルボキシル末端約150アミノ酸の領域は、TNFファミリーのメンバーと 顕著な相同性を示す。またこの領域は、TNFと同様、マトリックスメタロプロテアーゼ様の蛋白分解酵素により切リ出され、可溶型Fasリガンドを生ずる(3)。この可溶型Fasリガンドはマウスでは不活性であるが、ヒトでは細胞傷害活性を保持している。ヒト可溶型Fasリガンドは何らかの役割を担っている可能性があるが、膜型のFasリガンドに比べると細胞傷害活性が弱く、また傷害できる標的の種類も限られてくる(4)。従って、Fasリガンドの場合は、メタロプロテアーゼによる可溶化は活性を抑制する機構と考えられる。
|
図3 Fasと I 型TNF受容体のシグナル伝達機構 FasリガンドとFasの相互作用はアダプター分子FADDを介しカスパーゼ群に伝えられる。TNF受容体からのシグナルもアダプター分子群TRADD-FADD,
TRADD-RIP-RAIDDを介してやはりカスパーゼ群に伝えられる。RIPにはNF-κBの活性化を介してアポトーシスを抑制する働きもある。 |
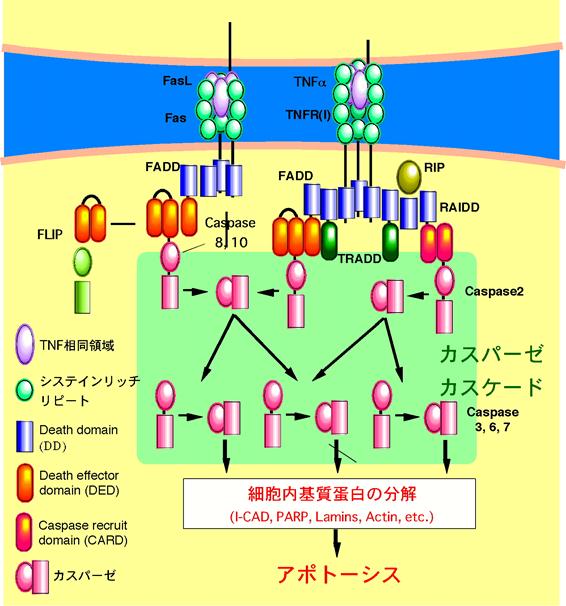
Fas(Apo-1/CD95)はTNF受容体ファミリーに属する細胞表面受容体で、Fasリガンドやアゴニスティックな抗Fas抗体が結合すると典型的なアポトーシスを誘導する(5-7)。細胞外領域にはTNF受容体ファミリーに特徴的なシステインリッチなドメインの繰り返し構造が3つ存在する(図3)。この部分にリガンドが結合し、受容体のホモ三量体化を誘導すると考えられている。FasとI型TNF受容体 (TNFR1) の細胞質領域には約70アミノ酸にわたり有為な相同性を示す領域が存在する。この領域は両分子の細胞傷害機能に重要などメインであるとことから、デスドメイン(death domain, DD)と呼ばれる(8, 9)。この領域にはアポトーシスのシグナル伝達に関るアダプター分子が結合する(後述)。
TNFファミリーに属するTRAIL (TNF-ralated apotosis-inducing
ligand) やTRANCE (TNF-ralated activation-induced
cytokine) には、これらのリガンドに結合するが細胞内シグナルを発しないおとり受容体が知られていたが、Fasリガンドに対しても可溶型のおとり受容体、Decoy Receptor 3 が発見された(10)。DcR3は4つのシステインリッチドメインを持ち、構造的にはFasよりもTRANCEのおとり受容体であるosteoprotegrinに近い。Fas遺伝子からもスプライシング亜型として可溶型Fasが生じる。これらの可溶型受容体はナチュラルなFasリガンド中和分子として働くと考えられる。
2.
Fasの細胞内シグナル伝達機構
現在までにFasのシグナル伝達機構の概要はほぼ明らかになっている (図3)(11)。Fasリガンドによる受容体の三量体化が引き金になって、FasのDDにはFADD
(Fas-associating protein with deth domain/ MORT1)と呼ばれる分子が結合する(12, 13)。TNFR1のDDにもTRADD (TNF receptor 1-associated death domain protein)を介してFADDやRIP (receptor interacting protein) が結合しうる。これらの分子はデス受容体と次に述べるカスパーゼ群との橋渡しをする分子であることから、アダプターと呼ばれる。これらのアダプター分子はやはりDDを持ち、DD-DD相互作用でデス受容体や他のアダプター分子と結合する。FADD のアミノ末端 (N末) には、カスパーゼ8 (FLICE/ MACH) およびカスパーゼ10 (Mch4) が結合する。カスパーゼ8や10のN末にはFADDのN末と共通のモチーフ(daeth
effector domain, DED) が二つ存在し、DED-DED相互作用でFADDと結合する。この他、RIPはRAIDDを介してカスパーゼ2に結合する(14)。RAIDDはDDとCARD (caspase recruit domain) をもち、DD-DD相互作用でRIPに結合し、CARD-CARD相互作用でCARDを持つカスパーゼ2と相互作用する。カスパーゼは不活性型のプロエンザイムとして産生され、自己消化あるいは他のカスパーゼなどにより切断されることにより活性化される。カスパーゼ2, 8, 10も不活性型で存在し、デス受容体複合体に取り込まれると自己消化により活性化されると考えられる。活性型のカスパーゼ8や10はカスパーゼ3, 6, 7などの下流カスパーゼを活性化する。さらに下流カスパーゼは様々な細胞内蛋白を切断することにより活性化あるいは不活性化し、アポトーシスに特徴的な変化を引き起こし、細胞を死に至らしめると考えられる。アポトーシスに特徴的な染色体DNAの断片化を引き起こすエンドヌクレアーゼ (CAD; caspase activated
DNase) は通常ナチュラルなインヒビターであるICAD (inhibitor of CAD, DFF45)
との複合体として存在するが、カスパーゼ3の作用でI-CADが分解されるとCADが活性化し、DNAの断片化を引き起こすことが明らかにされている(15, 16)。
ヘルペスウイルスなどの遺伝子にカスパーゼ 8, 10のようにDED領域を2つ持つタンパクをコードするものが存在する。この分子を細胞に発現させるとFasへのFLICEの結合が妨げられると同時に、Fasを介した細胞死も抑制されることから、viral FLICE-inhibitory
proteins (vFLIP) と呼ばれる(17)。Fasリガンドによる宿主細胞のアポトーシスを防ぐことは、ウイルスの増殖に有利に働くと考えられる。ヒトからも、DED領域を2つと、さらにカスパーゼ様の領域を持ちながら、カスパーゼとしての活性中心が存在しない分子が発見され、cellular FLIP (cFLIP), I-FLICE, FLAME-1, CASH, MRIT, CLARP, Casper など多くの名前で呼ばれている。しかしこの分子がvFLIPのようにアポトーシスを抑制するのか、それともカスパーゼ8, 10のようにアポトーシスのシグナルを伝達する分子なのかという点で、意見が真っ二つに割れている。
FasリガンドとFasがいったん結合すると、タンパク合成を止めてもアポトーシスは起こってしまう。すなわち、Fasのシグナル伝達分子は構成的に発現していることになる。Fasリガンドのシグナルは受容体直下でカスパーゼの活性化が起こるので、Bcl-2ファミリーの制御を受けない場合がある。 しかし、この直接的なカスパーゼの活性化がアポトーシスを誘導するに十分なレベルに達しないときは、カスパーゼ8がアポトーシス促進性Bcl-2ファミリーに属するBidを切断することによりBidのアポトーシス誘導能を高めたり、カスパーゼ3がアポトーシス抑制性のBcl-2を切断してアポトーシス促進型に転換したりすることにより、デスシグナルを増幅する回路があるらしい(18-20)。
II.
Fas-Fasリガンド系の生理的・病理的役割
1. FasとFasリガンドの発現
FasのmRNAはヒト及びマウスにおいて、ほとんどの組織、様々な細胞系列で広範に発現している。特に、胸腺、肺、心臓、肝臓、小腸で強く発現しており、逆に脳、精巣での発現は最も弱い。一方、FasリガンドmRNAは胸腺、脾臓、リンパ節などのリンパ系組織のほか腸、肺に発現している(21)。現在までに報告のあるFasリガンド発現細胞を表1にまとめた。リンパ球系の細胞では、活性化したT細胞とNKが発現している(22)。この他、好中球や樹状細胞がFasリガンドを発現するという報告もある。腸や肺は粘膜内リンパ組織に富む臓器であり、これらの組織で検出されたFasリガンドmRNAも結局はリンパ球に由来するものである可能性がある。また大腸上皮のPaneth細胞という特殊な細胞がFasリガンドを発現するという報告もある。マウスでは精巣のSertoli細胞や眼球の網膜側の非骨髄系細胞がFasリガンドを発現している。目や精巣は免疫系の監視から免れている (Immune previleged)ことが古くから知られているが、この現象は組織に発現したFasリガンドが浸潤してきた炎症細胞を殺してしまうためだと説明されている(23, 24)。この他、HIVウイルスに感染した単球、アルコール肝炎の患者の肝細胞、種々の癌細胞でFasリガンドの発現が報告されている。
|
表1 Fasリガンドの発現 ------------------------------------------------------------------------- リンパ・血液系細胞 活性化T細胞 ナチュラルキラー (NK) 細胞 単球 (HIV 感染) 好中球 樹状細胞 T-LGL白血病, NK-LGL白血病, NKリンパ腫 急性T細胞性白血病細胞 (抗がん剤誘導性) 非リンパ・血液系細胞 精巣セルトリ細胞 前眼膀の非骨髄由来細胞 肝細胞 (アルコール性肝炎) 腸管上皮Paneth細胞 甲状腺濾胞細胞 がん細胞 大腸癌、メラノーマ、肝癌 (抗がん剤誘導性)、乳癌 (ビタミンE誘導性)、肺癌 |
2. FasとFasリガンドの突然変異マウスが教えてくれたこと。
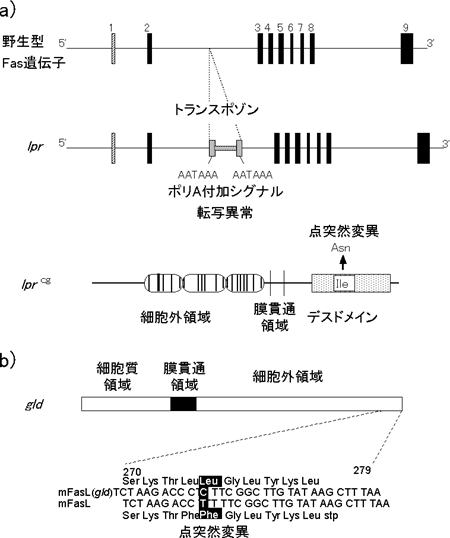
マウスFas遺伝子は19番染色体にマップされたが、同じ位置にlpr (lymphoproliferation) 突然変異遺伝子がマップされていた。このことをきっかけに、lpr はFasの突然変異であることが明らかにされた(図4)。lprマウスではFas遺伝子の第2イントロンにトランスポゾンの挿入があり、機能的なFas mRNAの発現が著しく低下している(25)。また、lprの対立遺伝子で、lpr と同様の異常をきたすlprcg突然変異遺伝子では、Fasの細胞質領域をコードするエクソン9 にイソロイシンをアスパラギンに置換するpoint mutationが存在し、このためFasの細胞死誘導活性が著しく減弱していることが示された(26)。一方、マウスFasリガンド遺伝子の座位を決めたところ、1番染色体のgld (generalized lymphoproliferative disease) 突然変異遺伝子の遺伝子座位と一致した(27)。さらに、gld 由来のFasリガンドcDNAにはコーデイング領域3'末近傍にチミジンからシトシンへの置換が存在し、結果として、gld 型FasリガンドはC末端から7番目のフェニルアラニンがロイシンに置換され、細胞死誘導活性を失っていることが判明した。
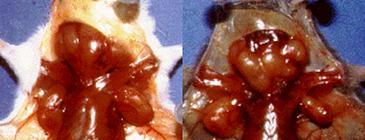
lprとgldは、ともに自然発生した劣性突然変異で、これらの変異遺伝子をホモで持つマウスは、生後数か月で、CD4もCD8も発現していないポリクローナルな異常T細胞が蓄積し、リンパ節腫や脾腫を引き起こす(図4c)(28)。また同じ時期に、抗DNA抗体などの自己抗体の産生が起こり、全身性の自己免疫疾患を発症する。このことからFasリガンドはリンパ球、特にT細胞のホメオスタシスと自己寛容の成立、維持に不可欠な役割を果たしていることが明らかになった。この発見の後、異常なT細胞の増加と自己免疫疾患を発症するヒト遺伝病でもFas遺伝子の突然変異が確認され、Fas-Fasリガンドシステムが、ヒトでも免疫系のホメオスタシスや自己寛容に重要な役割を果たしていることが明らかになった(29, 30)。
|
c)
|
図4 lprとgld;FasとFasリガンドの自然発生突然変異 a, b) lpr (a)およびgld突然変異(b)の構造。詳細は本文参照。 c) lpr (写真左)やgld (右)のホモ接合体マウスは、異常な表面形質をもつT細胞が著しく増加し、リンパ腫を発症する。この異常T細胞の増殖性は弱く、死なないために蓄積している。また、自己抗体産生にもとづく全身性の自己免疫疾患を引き起こす。 |
なぜFas-Fasリガンドシステムの異常は自己寛容の破綻を引き起こすのだろうか。活性化されたT細胞は、状況によっては抗原による再刺激を受けると、自分自身の発現したFasリガンドでアポトーシスを起こすことが知られている。このことから一つの仮説として、末梢の自己反応性T細胞は、通常自己抗原で刺激を受けるとFasリガンドを発現して自殺しており、このメカニズムが働かないlprやgldマウスでは、自己反応性T細胞が死なないために自己反応性B細胞の活性化を助け、自己免疫疾患を引き起こすのではないかという可能性が考えられている。しかし、我々の研究からlprマウスのT細胞にのみ野生型Fas遺伝子を発現させたトランスジェニックマウスでは、異常T細胞の蓄積によるリンパ腫は起こらなくなるが、自己抗体の産生や自己免疫疾患は余り抑制されないことが判明した(31)。従って、lprやgldマウスが自己免疫疾患を発症するのは、Fasリガンドによる自己反応性B細胞の除去が起こらないことがより本質的な原因ではないかと考えられる。ただし上述の結果は、Fasリガンドが自己反応性T細胞の排除に関与する可能性を否定するものではない。
3.
殺し屋T細胞 の武器;
細胞傷害性T細胞 (CTL) はウイルスに感染した細胞や癌細胞を殺す能力を持っている。このCTLによる標的細胞の細胞死もアポトーシスである。CTLの細胞傷害分子機構としては、CTLの分泌顆粒の中に存在するCa++依存性に標的細胞の細胞膜に穴を開けるパーフォリン (perforin) とこの穴から標的細胞に侵入してアポトーシスを誘導するグランザイム (granzyme) を介したものが知られていた(図5a)。しかし、CTLはCa++非存在下でも標的細胞を殺す場合があること、パーフォリンをほとんど発現していないCD4陽性T細胞も細胞傷害活性を示す事から、CTLにはパーフォリン/グランザイム系以外にも細胞傷害機構が存在すると考えられていた。このCTLの第2の細胞傷害機構がFasリガンドにより担われていることが明らかになった(図5b)。
|
図5 細胞傷害性T細胞のFasリガンド-Fas及びパーフォリン-グランザイムを介した細胞傷害機構a) パーフォリン/グランザイム依存性細胞傷害機構。パーフォリンはCTLの分泌顆粒に存在する補体成分C9に類似する分子量約70kDの蛋白質で、Ca++依存性に細胞膜に5〜20nmの孔を形成し、単独でも赤血球などを溶解する活性を有する。しかし実際のCTLでは、パーフォリンによる細胞膜の損傷だけでは細胞傷害の効率が低く、同じ分泌顆粒中に存在するグランザイムがパーフォリンの孔を通って標的細胞に侵入することが効率のよい細胞傷害に必要である。グランザイムは一群のセリンプロテアーゼで、グランザイムA〜HおよびTryptase-2、Met-ase-1の少なくとも10種類が知られている。この内、グランザイムBはカスパーゼ2,3,7〜10など を切断して活性化し、CTLによる標的細胞のアポトーシスに必須であるとされている。 b) Fasリガンド依存性細胞傷害機構。抗原受容体からのシグナルは、Fasリガンド遺伝子の転写を促進すると同時に、分泌顆粒に蓄えられていたFasリガンド分子を細胞表面に移動させる。細胞表面に発現したFasリガンドは標的細胞のFasに作用してアポトーシスを誘導する。 |
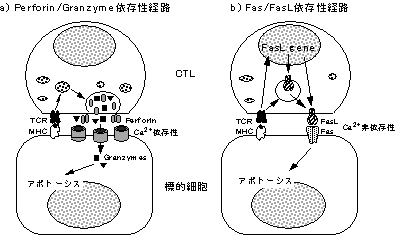
FasリガンドmRNAの発現はT細胞受容体からの抗原刺激で誘導される(21)。また、分泌顆粒にプールされていたFasリガンドは抗原刺激で誘導される脱顆粒によって、速やかに標的細胞との接触面に移動する(32)。一方、いったん細胞表面に出現したFasリガンドは、比較的短時間でメタロプロテアーゼにより細胞外領域が切断される。切断された細胞外領域は可溶型Fasリガンドとなるが、マウスのそれはほとんど細胞傷害活性を示さず、ヒトでもかなり活性が減弱する(4, 33)。このため、Fasリガンドによる細胞傷害もある程度抗原特異性を示す。しかしFasを強く発現する細胞に対しては抗原非特異的な細胞傷害活性を示す。
4.
Fasリガンドは両刃の刃
FasやFasリガンドの欠損が全身性自己免疫疾患や異常T細胞の増加の原因になることは既に述べたが、FasやFasリガンドの過剰発現も疾患の原因になる可能性がある。
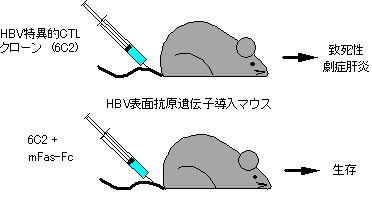
我々は、Fasを介して細胞死を誘導する活性を持つ抗マウスFas抗体をマウスに投与すると、大量の肝細胞が一度にアポトーシスを起こし、3時間以内にマウスを死滅させることを発見した(34)。この結果は、肝実質細胞がFasリガンドに感受性が高いことを示す。ウイルス性肝炎などで、CTLの病理的な関与が示唆されており、FasリガンドがCTLのエフェクター分子として働いていることを考え合わせると、肝細胞上のウイルス抗原を認識したCTLがFasリガンドを発現し、これが肝細胞上のFasと結合し、肝細胞にアポトーシスを誘導するというメカニズムが、劇症肝炎を引き起こしている可能性が考えられる。実際、劇症肝炎の動物モデルを利用し、Fasリガンドに対する中和作用を示すFasの細胞外領域とヒト免疫グロブリンGのFc領域の融合蛋白が、劇症肝炎に対する治療効果を示すことが明らかになった (図6) (35)。また、骨髄移植や臓器移植で問題となる移植片対宿主(GVH)病などに於ても、Fasリガンドが病理的に働いていることが示されている。我々の作成したマウスFasリガンドに対する中和抗体は、マウスのGVH病モデルで一定の治療効果を示す(36)ことから、今後様々な疾患に対しFasリガンドの機能制御に基づいた治療法を検討してみる必要があると考えている。
|
図6 細胞傷害性T細胞誘導致死性劇症肝炎のFas-Fcによる治療モデル。 B型肝炎ウイルス(HBV)の表面抗原遺伝子をアルブミンプロモーターの制御下に肝臓特異的に発現させたマウスに、HBV特異的CTLを移入すると、劇症肝炎を引き起こし三日以内に死亡する。このモデルで、Fasリガンドに対する中和作用を持つFas-FcをCTLと同時に投与すると、マウスは生き延びることが出来る。 |
5.
Fasリガンドと炎症
ネクローシス(壊死)ではプロテアーゼなどを含む細胞内容物が漏出し、周囲の細胞を傷害したり、炎症を引き起こすと考えられている。これに対し、アポトーシスを起こした細胞は、内容物が漏出する前に貪食細胞や周囲の細胞に吸収されてしまうため炎症は誘導しないというのがこれまでの通説であった。しかし、Fasリガンドと炎症の関係については、FasリガンドがT細胞ではなく組織や癌細胞側に発現した場合に、炎症を抑制するという報告と誘導するという報告が入り乱れ、ホットな議論になっている。
脳、眼、精巣などの組織では炎症が起こりにくいことから、免疫系の攻撃を特権的に免れる組織という意味で、免疫特権組織 (immune privileged organ) と呼ばれる(37)。炎症は本来、侵入してきた異物やがん細胞を排除するための生体防御的反応ではあるが、過剰な炎症反応は往々にして自己組織を傷害し、機能障害を引き起こすため、これらの組織では炎症が起きにくい機構が発達したものと考えられる。最近、眼や精巣にはFasリガンドが発現しており、浸潤してきた炎症細胞にアポトーシスを誘導することによって炎症を抑制していることが示された(37)。ある種のがん細胞もFasリガンドを発現していることから、がん細胞が免疫系の攻撃を回避する一つの手段としてFasリガンドを利用している可能性も示唆された(38)。一方、遺伝子導入によってがん細胞や正常組織に異所性にFasリガンドを発現させると、むしろ炎症が惹起されて拒絶されたり組織破壊が起こることが見出された(39, 40)。
|
|
図7 Fasリガンドによるアポトーシスと炎症 IL-1βの不活性型前駆体を発現している細胞にFasリガンドを作用させると、カスパーゼの活性化によりアポトーシスと同時にプロセシングによるIL-1βの活性化と放出が誘導され、炎症が惹起されると考えられる。リード 死ぬことも増殖と同様に重要な細胞の機能である。高等動物は細胞に死を促すサイトカイン=デス因子を作り出した。デス因子の代表的存在であるFasリガンドのシグナル伝達機構と生物学的役割とは。 |
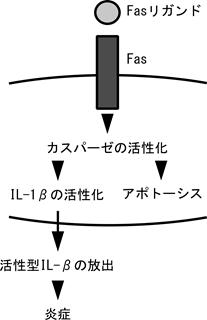
なぜFasリガンドは炎症を誘導するのだろうか。Fasリガンドはカスパーゼの活性化を介してアポトーシスを誘導することは既に述べたが、カスペース1はもともと炎症性サイトカインであるIL-1βを不活性な前駆体から活性型に転換する酵素として知られていた。従って前駆体IL-1βを発現している細胞にアポトーシスが誘導されれば、IL-1βが活性化し炎症を促進する可能性がある。実際、最近の我々の研究から、Fasリガンドを発現した癌細胞は、好中球に富む炎症性浸潤細胞にアポトーシスと同時にIL-1βの前駆体から活性型への転換と放出を誘導し、このIL-1βによって炎症がさらに増強されるらしいことが判明した (図7) (41)。(誤解がないように付け加えれば、ここで述べたような機構が働くのはアポトーシスを起こした細胞がIL-1β前駆体を発現している場合だけである。)
この結果から考えると、Fasリガンドを異所性に発現させると、体中を循環している好中球や弱い炎症反応で集まった炎症細胞などがその場でアポトーシスを起こし、活性型のIL-1βを放出して炎症を誘導、促進するものと考えられる。前述のようにカスパーゼの一部が炎症性サイトカインの活性化を担っていることを考えれば、アポトーシスと炎症の間に排他的な関係よりはむしろ密接な相互関係があるだろうことは想像に難くない。興味深いことに、Fasリガンド刺激によるIL-1βの産生は、カスパーゼ阻害剤により抑制されるが、カスパーゼ1遺伝子を破壊したマウス由来の炎症細胞を用いても起こる。すなわち、この場合のIL-1βのプロセシングはカスパーゼ1以外のカスパーゼを介して起こりうると考えられる。我々は劇症肝炎などの炎症性疾患においてもFasリガンドによるアポトーシスが炎症の引き金を引いている可能性を示唆してきた。アポトーシスと炎症の関係を見直すと、アポトーシスと疾患の関係について新たな側面が見えてくるかもしれない。なぜ免疫特権組織ではFasリガンンドの発現により炎症が抑えられるのかは不明であるが、免疫特権組織では、炎症細胞のアポトーシスによりIL-1βが放出されても、その活性を抑制するメカニズムがあるのかもしれない。
おわりに
Fasリガンドなどのデス因子の存在、すなわち「細胞死がサイトカインによって制御されている」という事実は、サイトカイン研究者ばかりでなく多くの研究者にアポトーシスの機能的重要性を印象付け、アポトーシス研究の現在の隆盛の一つの起爆剤になった。また、in vitroでサイトカインという生理的な物質で極めて直接的にアポトーシスを誘導することが出来るようになり、アポトーシス分子機構の研究の大きな推進力にもなている。Fasリガンドのシグナル伝達機構は、細胞外の刺激から最終的な死の執行までひととおりつながった。しかし、それを正・負に修飾する因子はFLIP以外にも未発表のものも含め色々と存在するようであり、どのような場合にどのような制御機構が働いているのかということまで含めると、全容の解明には未だしばらく時間がかかりそうである。一方、Fasリガンドの役割としては、リンパ球のホメオスタシスや自己寛容、CTLの細胞傷害因子としての役割などが明らかにされて来たが、最後に述べた炎症との関りなど、新しい役割も見出されている。Fasリガンドが好中球に対するケモカインとして働くという報告もある。疾患との関りについても、ここで述べた全身性の自己免疫疾患や劇症肝炎、GVHD、癌以外にも局所型の自己免疫疾患、エイズなど様々な現代の難病との関りが示唆されている。今後は、Fasリガンドを利用したり、逆にその機能を抑制することによりこれらの疾患を治療する方法が検討されていくことになると思われる。デス因子全般に話を広げると、Fasリガンドの後にもTNF-related apoptosis-inducing
ligand (TRAIL/Apo2L) 、Apo3Lが発見され、それらの受容体Death receptor (DR)3, DR4, DR5が発見されている。これらの分子の生理的役割は未だ充分に解明されていない。さらに、CAR1やDR6などデス因子受容体と考えられるがリガンドが未発見の分子もあり、まだまだデス因子に関る新しい分子や生理的役割の発見が相次ぐと思われる。発生や老化に関るデス因子は存在するだろうか。急速に進展しているとはいえ、デス因子の研究はまだまだ始まったばかりである。
文献
1. Suda, T., Takahashi,
T., Golstein, P., Nagata, S. : Cell 75, 1169-1178 (1993).
2. Suda, T., Nagata, S. :
J. Exp. Med. 179, 873-878 (1994).
3. Tanaka, M., Suda, T.,
Haze, K., Nakamura, N., Sato, K., Kimura, F., Motoyoshi, K., Mizuki, M.,
Tagawa, S., Ohga, S., Hatake, K., Drummond, A.H., Nagata, S. : Nature Medicine
2, 317-322 (1996).
4. Suda, T., Hashimoto,
H., Tanaka, M., Ochi, T., Nagata, S. : J Exp Med 186, 2045-2050 (1997).
5. Yonehara, S., Ishii,
A., Yonehara, M. : J. Exp. Med. 169, 1747-1756 (1989).
6. Itoh, N., Yonehara, S.,
Ishii, A., Yonehara, M., Mizushima, S., Sameshima, M., Hase, A., Seto, Y., Nagata,
S. : Cell 66, 233-243 (1991).
7. Trauth, B.C., Klas, C.,
Peters, A.M.J., Matzuku, S., M嗟ler, P., Falk, W., Debatin, K.-M., Krammer, P.H.
: Science 245, 301-305 (1989).
8. Itoh, N., Nagata, S. :
J. Biol. Chem. 268, 10932-10937 (1993).
9. Tartaglia,
10. Pitti, R.M., Marsters, S.A.,
Lawrence, D.A., Roy, M., Kischkel, F.C., Dowd, P., Huang, A., Donahue, C.J.,
Sherwood, S.W., Baldwin, D.T., Godowski, P.J., Wood, W.I., Gurney, A.L.,
Hillan, K.J., Cohen, R.L., Goddard, A.D., Botstein, D., Ashkenazi, A. : Nature
396, 699-703 (1998).
11. Nagata, S. : Cell 88, 355-365
(1997).
12. Chinnaiyan, A.M., O'Rourke, K.,
Tewari, M., Dixit, V.M. : Cell 81, 505-512 (1995).
13. Boldin, M.P., Varfolomeev, E.E.,
Pancer, Z., Mett, I.L., Camonis, J.H., Wallach, D. : J. Biol. Chem. 270,
7795-7798 (1995).
14. Duan, H., Dixit, V.M. : Nature 385,
86-89 (1997).
15. Enari, M., Sakahira, H., Yokoyama,
H., Okawa, K., Iwamatsu, A., Nagata, S. : Nature 391, 43-50 (1998).
16. Sakahira, H., Enari, M., Nagata, S.
: Nature 391, 96-99 (1998).
17. Thome, M., Schneider, P., Hofmann,
K., Fickenscher, H., Meinl, E., Neipel, F., Mattmann, C., Burns, K., Bodmer,
J.L., Schroter, M., Scaffidi, C., Krammer, P.H.,
18. Li, H., Zhu, H., Xu, C.J., Yuan, J.
: Cell 94, 491-501 (1998).
19. Luo, X., Budihardjo,
20. Cheng, E.H., Kirsch, D.G., Clem,
R.J.,
21. Suda, T.,
22. Arase, H., Arase, N., Saito, T. : J
Exp Med 181, 1235-1238 (1995).
23. Bellgrau, D., Gold, D., Selawry,
H.,
24. Griffith, T.S., Brunner, T.,
Fletcher, S.M., Green, D.R.,
25. Adachi, M., Watanabe-Fukunaga, R.,
Nagata, S. : Proc. Natl. Acad. Sci. USA 90, 1756-1760 (1993).
26. Watanabe-Fukunaga, R., Brannan,
C.I., Copeland, N.G., Jenkins, N.A., Nagata, S. : Nature 356, 314-317 (1992).
27. Takahashi, T., Tanaka, M., Brannan,
C.I., Jenkins, N.A., Copeland, N.G., Suda, T., Nagata, S. : Cell 76, 969-976
(1994).
28. Cohen, P.L., Eisenberg, R.A. : Ann.
Rev. Immunol. 9, 243-269 (1991).
29. Fisher, G.H., Rosenberg, F.J.,
Straus, S.E., Dale, J.K., Middleton, L.A., Lin, A.Y., Strober, W., Lenardo,
M.J., Puck, J.M. : Cell 81, 935-946 (1995).
30. Rieux-Laucat, F., Le, D.F., Hivroz,
C., Roberts, I.A., Debatin, K.M., Fischer, A., de, V.J. : Science 268,
1347-1349 (1995).
31. Fukuyama, H., Adachi, M., Suematsu,
S., Miwa, K., Suda, T., Yoshida, N., Nagata, S. : J Immunol 160, 3805-3811
(1998).
32. Bossi, G., Griffiths, G.M. : Nat
Med 5, 90-96 (1999).
33. Tanaka, M., Itai, T., Adachi, M.,
Nagata, S. : Nat Med 4, 31-36 (1998).
34. Ogasawara, J., Watanabe-Fukunaga, R.,
Adachi, M., Matsuzawa, A., Kasugai, T., Kitamura, Y., Itoh, N., Suda, T.,
Nagata, S. : Nature 364, 806-809 (1993).
35. Kondo, T., Suda, T., Fukuyama, H.,
Adachi, M., Nagata, S. : Nat Med 3, 409-413 (1997).
36. Miwa, K., Hashimoto, H., Yatomi,
T., Nakamura, N., Nagata, S., Suda, T. : Int. Immunol. in press (1999).
37.
38. Hahne, M., Rimoldi, D., Schroter,
M., Romero, P., Schreier, M., French, L.E., Schneider, P., Bornand, T.,
Fontana, A., Lienard, D., Cerottini, J., Tschopp, J. : Science 274, 1363-1366
(1996).
39. Seino, K., Kayagaki, N., Okumura,
K., Yagita, H. : Nat Med 3, 165-170 (1997).
40. Kang, S.M., Schneider, D.B., Lin,
Z., Hanahan, D., Dichek, D.A., Stock, P.G., Baekkeskov, S. : Nat Med 3, 738-743
(1997).
41. Miwa, K., Asano, M., Horai, R.,
Iwakura, Y., Nagata, S., Suda, T. : Nat Med 4, 1287-1292 (1998).