Research Progress of the Center for the Development of Molecular
Target Drugs (2003-2005)
Suda’s group
A) Molecular mechanism of Fas
ligand-induced inflammation
Fas ligand (FasL) is a
prototype of death factor that induces apoptosis by binding to its receptor,
Fas. We previously demonstrated that anti-Fas ligand antibody administration
ameliorates various inflammatory diseases, and prevent
chronic-hepatitis-induced hepatic cancer development using animal models. We
also demonstrated that Fas ligand-induced caspase activation in neutrophils
causes not only apoptosis but also proteolytic maturation and release of IL-1b,
which in turn induces massive neutrophil infiltration in vivo. In the last three
years, we further demonstrated that Fas ligand induces production of various
cytokines including IL-1b, IL-6, IL-17, IL-18, IL-23, KC, and Mip2 in mice. Fas
ligand induces IL-23 production in dendritic cells at the mRNA level in a
cell-autonomous manner. The IL-23 then induces IL-17 production in T cells
synergistically with IL-1b. We also found that Fas ligand induces IL-8
production in human embryonic kidney (HEK) 293 cells in a manner dependent on
the NF-kB
activation. The Fas ligand-induced NF-kB activation is a cell-autonomous
response and requires FADD and caspase-8.
B) Molecular mechanisms of activation and regulation of ASC-mediated inflammation.
ASC
is an adaptor molecule that mediates apoptotic and inflammatory signals from
several Apaf-1-like molecules, including CARD12/Ipaf, PYPAF1/cryopyrin, PYPAF5,
PYPAF7, and NALP1. ASC is also implicated in tumor suppression, because the ASC
gene expression is suppressed in various cancer cells. We have established an
experimental system in which muramyl dipeptide, the bacterial component recognized by
another Apaf-1-like molecule, Nod2, induced an interaction between a
CARD12-Nod2 chimeric protein and ASC, and elicited cell-autonomous NF-kB activation leading to IL-8
production. Using this system, we demonstrated that caspase-8 plays an
essential role in ASC-mediated NF-kB activation. On the other hand, we have
found that some members of Apaf-1-like proteins such as PYNOD, PYPAF2 and
PYPAF3 inhibit ASC-mediated NF-kB activation and or caspase-1-mediated
proteolytic maturation of IL-1b.
Molecular mechanisms of
Fas ligand-induced inflammation.
M. Umemura, T. Kawabe, H. Kidoya, K. Shudo, M. Fukui, M.
Asano, Y. Iwakura, G. Matsuzaki,A. Yahagi, R. Imamura, and T. Suda
Fas ligand (FasL) has been
well characterized as a death factor. However, recent studies revealed that
ectopic expression of FasL induces inflammation associated with massive
neutrophil infiltration. We previously demonstrated that the neutrophil
infiltration-inducing activity of FasL is partly dependent on but partly
independent of IL-1b. Therefore,
we investigated the cytokine profile of peritoneal lavage fluid obtained from
mice that received intraperitoneal injections of FFL, a FasL-expressing tumor
cell line. We found that FFL injection caused a marked increase of not only
IL-1b but also
IL-6, IL-17, IL-18, KC/chemokine CXC ligand 1, and macrophage
inflammatory protein (MIP)-2, but not of IL-1a,
IFN-g, TGF-b, or TNF-a. Among
cells transfected to express individually IL-1b,
IL-6, IL-17, or IL-18, only those expressing IL-1b
and IL-17 induced neutrophil infiltration. Co-administration
of the anti-IL-17 antibody with FFL diminished the peritoneal KC levels and
neutrophil infiltration in IL-1-deficient mice. In addition, the expression of
IL-17 by the tumor cells inhibited tumor growth in wild-type and nude mice.
These results suggest that IL-17 is involved in FasL-induced inflammation and
tumor rejection in the absence of IL-1b.
Then,
we investigated the mechanism of the FasL-induced IL-17 production. We found
that the culture supernatant of mouse resident peritoneal exudate cells (PEC)
cocultured with FFL cells induced IL-17 production in freshly isolated resident
PEC. Anti-IL-1b
Ab strongly inhibited the IL-17-inducing activity. However, recombinant IL-1b by itself induced only weak
IL-17 production. Intriguingly, anti-IL-12 Ab but not an IL-15 neutralizing
agent, IL15R-Fc, strongly inhibited the FasL-induced IL-17-inducing activity.
IL-23, which shares the p40 subunit with IL-12, but not IL-12 itself, induced
IL-17 production synergistically with IL-1b in resident PEC. FasL induced
the production of IL-23 in PEC in vivo
and in vitro, and IL-17 production
following the i.p. injection of FFL cells was severely impaired in p40-/- mice,
indicating that IL-23 plays an important role in the FasL-induced IL-17
production. FFL also induced the production of IL-23 in bone marrow- or
PEC-derived dendritic cells. Finally, FasL induced only weak p40 production in
a mixture of p40-/- and Fas-/- dendritic cells, indicating that FasL induces
IL-23 production in dendritic cells mainly in a cell-autonomous manner.
1.
Umemura, M., et al., Int. Immunol., 16:1099-108, 2004
2.
Kidoya, H., et al., J. Immunol., 175:8024-31, 2005
Molecular Mechanism of Fas ligand-induced IL-8 Production
R.
Imamura, N. Matsumoto, M. Hasegawa, K. Konaka, and T. Suda
It has been believed that apoptosis does
not induce inflammation. However, there are remarkable similarities between the
molecular mechanisms of apoptosis and inflammation. Fas (CD95) is not an
exception and recent studies revealed that Fas ligand (FasL)-Fas system
possesses inflammatory activity. We recently found that FasL induces production
of the inflammatory chemokine IL-8 in human cell lines and FasL-induced NF-kB and AP-1 activation is
required for this IL-8 production. Our further analyses revealed that the death
domain of Fas, FADD, and caspase-8, which are essential for the apoptosis
signaling, are required for both NF-kB and AP-1 activation by FasL. However,
rsponses of NF-kB
and AP-1 activation are independent of each other. In the NF-kB signaling pathway, we also
showed that TRADD and RIP, which are essential for the TNF-a-induced NF-kB activation, were not
involved in the FasL-induced NF-kB activation and CLARP/FLIP inhibited the
FasL- but not the TNF-a-induced NF-kB activation. More
interestingly, our results revealed that enzymatic activity of caspase-8 is
required for both NF-kB and AP-1 activation induced by FasL.
Further characterization of these pathways will help us to understand and,
hopefully, to control the FasL-induced inflammation. (Imamura, R. et al., J.
Biol. Chem., 279:46415-46423, 2004)
Figure
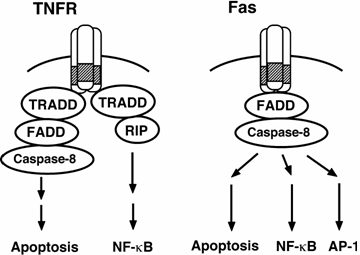 In the case of FasL-Fas
system, contrary to TNF-a-TNFR system, signaling pathway of
apoptosis and transcription factor activation separates downstream of caspase-8
and enzymatic activity of caspase-8 is required for both pathways. Current our
goal is identification of targets (substrates) of caspase-8, which are
important for FasL-induced NFkB and/or AP-1 activation.
In the case of FasL-Fas
system, contrary to TNF-a-TNFR system, signaling pathway of
apoptosis and transcription factor activation separates downstream of caspase-8
and enzymatic activity of caspase-8 is required for both pathways. Current our
goal is identification of targets (substrates) of caspase-8, which are
important for FasL-induced NFkB and/or AP-1 activation.
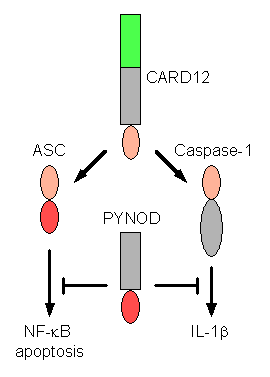
PYNOD, a Novel Apaf-1/CED4-like Protein is an Inhibitor of ASC and Caspase-1
Y. Wang, M. Hasegawa, R. Imamura, T. Kinoshita, C. Kondo,
K. Konaka, and T. Suda
Recently, a large subfamily of
nucleotide-binding and oligomerization domain-containing proteins that have an
N-terminal pyrin-like domain and C-terminal leucine-rich repeats has been
described. In this study, we identified PYNOD, a novel member of this family
that lacks the leucine-rich repeats. We found that human PYNOD mRNA is
expressed in various tissues and at high levels in heart, skeletal muscle, and
brain. It is also expressed in various cell lines, including haematopoietic
cell lines. PYNOD oligomerizes and binds to ASC, an adaptor protein that plays
a role in apoptotic and inflammatory signal transduction, and to caspase-1 and
IL-1b.
PYNOD inhibits ASC-mediated NF-kB activation and apoptosis, and
caspase-1-mediated IL-1b maturation, and it does so in the
presence and absence of constitutively active mutants of CARD12 and PYPAF1,
which are enhancers of these processes. Thus, PYNOD is a novel regulator of
apoptosis and inflammation. (Wang, Y. et al., Int. Immunol. 16:777-86, 2004)
|
Figure PYNOD is a member of Apaf-1-like proteins and inhibits ASC-mediated NF-kB activation and apoptosis, and caspase-1-mediated IL-1b activation. |
|
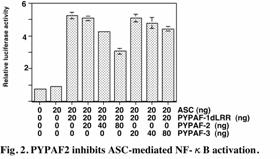
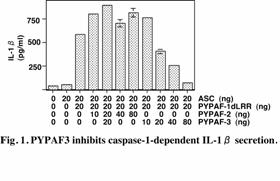
PYPAF3, a PYRIN-containing
Apaf-1-like protein, is a feedback regulator of caspase-1-dependent
interleukin-1b secretion
T.
Kinoshita, Y. Wang, M. Hasegawa, R. Imamura, T. Suda
PYPAF3 is a member of the
PYRIN-containing apoptotic protease-activating factor-1-like proteins (PYPAFs) that
are thought to function in inflammatory signaling pathways. Among the members
of this family, PYPAF1, PYPAF5, PYPAF7, and NALP1 have been shown to induce
caspase-1-dependent interleukin-1b secretion and NF-kB activation in the presence
of the adaptor molecule ASC. On the other hand, we recently identified PYNOD,
another member of this family, as a suppressor of these responses. In this
study, we showed that PYPAF3 is the second member that inhibits
caspase-1-dependent interleukin-1b secretion (Fig. 1) and that PYPAF2 does
not inhibit this response, but rather inhibits the ASC-mediated NF-kB activation (Fig. 2). Both PYPAF2 and PYPAF3 mRNAs are broadly
expressed in a variety of tissues; however, neither is expressed in skeletal
muscle, and only PYPAF2 mRNA is expressed in heart and brain. They are also
expressed in many cell lines of both haematopoietic and non-haematopoietic
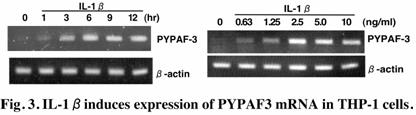
lineages. Stimulation of monocytic THP-1 cells with lipopolysaccharide or
interleukin-1b
induced PYPAF3 mRNA expression (Fig. 3). Furthermore, the stable expression of
PYPAF3 in THP-1 cells abrogated the cells’ ability to produce interleukin-1b in response to
lipopolysaccharide. These results suggest that PYPAF3 is a feedback regulator
of interleukin-1b
secretion. Thus, PYPAF2 and PYPAF3, together with PYNOD, may constitute an
anti-inflammatory subgroup of PYPAFs. (Kinoshita, T. et al., J. Biol. Chem. 280:21720-5, 2005)
ASC-mediated NF-kB Activation Leading to IL-8 Production Requires Caspase-8 and Is Inhibited by CLARP
M.
Hasegawa, R. Imamura, T. Kinoshita, N. Matsumoto, J. Masumoto, N. Inohara,
and T. Suda
ASC is an adaptor molecule
that mediates apoptotic and inflammatory signals from several Apaf-1-like
molecules, including CARD12/Ipaf. ASC is also implicated in tumor suppression,
because the ASC gene expression is suppressed in various cancer cells. To
characterize the signaling pathway mediated by ASC, we established cell lines
in which muramyl dipeptide, the bacterial component recognized by
another Apaf-1-like molecule, Nod2, induced an interaction between a
CARD12-Nod2 chimeric protein and ASC, and elicited cell-autonomous NF-kB activation. This response
required caspase-8, and was suppressed by CLARP/FLIP, an inhibitor of
caspase-8. The catalytic activity of caspase-8 was required for the
ASC-mediated NF-kB
activation when caspase-8 was expressed at an endogenous level, although it was
not essential when caspase-8 was overexpressed. In contrast, FADD, the adaptor
protein linking Fas and caspase-8, was not required for this response.
Consistently, ASC recruited Caspase-8 and CLARP but not FADD and Nod2 to its
speck-like aggregates in cells. Finally, muramyl
dipeptide induced
IL-8 production in MAIL8 cells. These results are the first to indicate that
caspase-8 plays an important role in the ASC-mediated NF-kB activation, and that the
ASC-mediated NF-kB
activation actually induces physiologically relevant gene expression. (Hasegawa
M, et al., J. Biol. Chem. 280: 15122-30, 2005.)
|
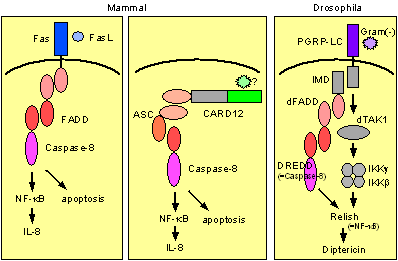
Figure DREDD, the drosophila homolog of caspase-8 has been shown to play an important role in NF-kB activation. In mammals, caspase-8 also plays an important role in Fas ligand-induced and ASC-mediated NF-kB activation. |
|
Publications
1. Kidoya, H., Umemura, U., Kawabe, T.,
Matsuzaki, G., Yahagi, A., Imamura, R., and Suda, T. (2005) Fas ligand induces
cell-autonomous IL-23 production in dendritic cells, a mechanism for Fas
ligand-induced IL-17 production. J. Immunol. 175:8024-31.
2. Kinoshita, T., Wang, Y., Hasegawa, M.,
Imamura, R., and Suda, T. (2005) PYPAF3, a PYRIN-containing APAF-1-like
protein, is a feedback regulator of caspase-1-dependent interleukin-1b
secretion. J. Biol. Chem. 280:21720-5
3. Hasegawa M, Imamura R, Kinoshita T,
Matsumoto N, Masumoto J, Inohara N, and Suda T. (2005) ASC-mediated NF-kB
activation leading to interleukin-8 production requires caspase-8 and is
inhibited by CLARP. J. Biol. Chem. 280: 15122-30.
4. Suda, T. (2005) Physiological and
pathological roles of apoptosis.
5. Imamura, R., Konaka, K., Matsumoto, N.,
Hasegawa, M.,
6. Umemura, M., Kawabe, T., Shudo, K.,
Kidoya, H.,
7. Wang, Y., Hasegawa, M., Imamura, R.,
Kinoshita, T., Kondo, C., Konaka, K., Suda, T. (2004) PYNOD, a novel
Apaf-1/CED4-like protein is an inhibitor of ASC and caspase-1. Int. Immunol.
16:777-86.
8. Nakamoto Y, Suda T, Momoi T, and Kaneko
S. (2004) Different procarcinogenic potentials of lymphocyte subsets in a
transgenic mouse model of chronic hepatitis B. Cancer Res. 64:3326-3333
9. Uekita, T., Gotoh,
10.
11. Tachiiri, A.,
Imamura, R., Wang, Y.,