General Summary of
Division of Immunology and Molecular
Biology (2006-2008)
In the last three years, our study has been
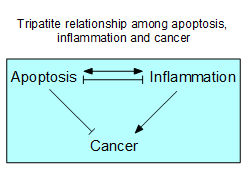
aiming to clarify the tripartite relationship among apoptosis,
inflammation and cancer. It is generally believed that apoptosis is an
important process to prevent oncogenesis, and that inflammation is a causative
factor for cancer development. In terms of the relationship between apoptosis
and inflammation, it has been said that apoptosis does not induce inflammation.
However, recent studies demonstrated that apoptosis and inflammation uses many
common protein molecules for signal transduction. For example, Fas ligand, an
apoptosis inducing factor, was found to be a potent inducer of inflammation in
vivo. Furthermore, we previously discovered that in a mouse model of
chronic
|
hepatitis that eventually develops hepatic cancer, administration of a
neutralizing antibody against Fas ligand suppressed the hepatic
inflammation and cancer development. Based on these results, we are investigating
the functions of proteins (including Fas ligand) that play a role at the
crossroad of apoptosis and inflammation, aiming to discover new findings useful
for cancer therapy. |
|
A) Study for the molecular mechanism of
the inflammatory activity of Fas ligand.
Fas ligand is famous as a death factor;
however, transplantation of Fas ligand-expressing cells into the peritoneal cavity
of a mouse induces peritonitis associated with massive neutrophil infiltration.
In addition, Fas ligand induces production of IL-8, a chemokine for
neutrophils, in the human embryonic kidney (HEK)-293 cell line. Our previous
study revealed that NF-kB plays an important role in the Fas
ligand-induced IL-8 production and that caspase-8 is involved in this response.
This time, we further demonstrated that AP-1 is also required for optimal IL-8
production, and that caspase-8 and JNK play essential roles in this response.
B) Study for the molecular mechanism of
ASC-mediated apoptosis
ASC was originally discovered as a protein that forms a large
aggregate in an apoptotic HL-60 human leukemia cell treated with a chemotherapeutic
drug. This protein was independently discovered as a product of the gene called
TMS1 whose expression in various human cancer tissues were suppressed by DNA
methylation. It has been also reported that the expression of ASC is controlled
by p53 tumor suppressor, and that ASC is involved in etoposide-induced
apoptosis of tumor cells. These results suggest that ASC is a novel tumor suppressor.
In addition, ASC was recently identified as an adaptor protein that mediates inflammatory
and apoptotic signals from some members of the NLR family (such as cryopyrin
and CARD12) that function as cytoplasmic sensors for pathogens and activate
cellular innate immune responses. The molecular mechanism of ASC-mediated
apoptosis has been controversially reported. Initially, it was reported to be
caspase-9 dependent; however, this notion was recently challenged. Therefore, we
have investigated that molecular mechanism of ASC-mediated apoptosis and
clearly demonstrated that caspase-8 plays an important role for this response. Just
like the extrinsic pathway of apoptosis that is initiated by a death factor,
ASC-mediated apoptosis in type-2 cells depends on proteolytic activation of Bid
by caspase-8.
Recently, we also discovered that ASC
mediates necrotic cell death under some conditions. Our current study is aiming
to clarify the molecular mechanism of ASC-mediated necrosis. We also
investigate possible use of ASC as a molecular target for cancer therapy.
C) The regulatory mechanisms of NLR
proteins
Several members of NLR proteins functions (including cryopyrin) as
cytoplasmic sensors for pathogens and activator for apoptosis and inflammation
as described above. However, we previously discovered other members of NLRs
such as PYNOD, PYPAF2 and PYPAF3 function as negative regulator for ASC and
caspase-1. This time, we have searched for cryopyrin-binding proteins using the
yeast-two hybrid system. As a result, we have identified FAF1 as a novel
cryopyrin-binding protein, and found that FAF1 inhibits cryopyrin-mediated NF-kB activation. In addition, we have established PYNOD-transgenic
mice, and we are now investigating the in vivo functions of PYNOD.
Caspase-8-and
JNK-dependent AP-1 activation is required for Fas ligand-induced IL-8 production
Norihiko
Matsumoto, Ryu Imamura, and
Despite a dogma that apoptosis does not induce
inflammation, Fas ligand (FasL), a well-known death factor, possesses
pro-inflammatory activity. For example, FasL induces nuclear factor kB (NF-kB) activity and interleukin 8 (IL-8)-production by
engagement of Fas in human cells. Here, we found that a dominant negative
mutant of c-Jun, a component of the activator protein-1 (AP-1) transcription
factor, inhibits FasL-induced AP-1 activity and IL-8 production in HEK293
cells. Selective inhibition of AP-1 did not affect NF-kB activation and vice-versa, indicating that their
activations were not sequential events. The FasL-induced AP-1 activation could
be inhibited by deleting or introducing the lymphoproliferation (lpr)-type
point mutation into the Fas death domain (DD), knocking down the Fas-associated
DD protein (FADD), abrogating caspase-8 expression with small interfering RNAs
(siRNAs), or using inhibitors for pan-caspase and caspase-8 but not caspase-1
or caspase-3. Furthermore, wild-type, but not a catalytically inactive mutant, of
caspase-8 reconstituted the FasL-induced AP-1 activation in caspase-8-deficient
cells. Fas ligand induced the phosphorylation of two of the three major
mitogen-activated protein kinases (MAPKs): extracellular signal-regulated
kinase (ERK) and c-Jun N-terminal kinase (JNK) but not p38 MAPK. Unexpectedly,
an inhibitor for JNK but not for MAPK/ERK kinase inhibited the FasL-induced
AP-1 activation and IL-8 production. These results demonstrate that
FasL-induced AP-1 activation is required for optimal IL-8 production, and this
process is mediated by FADD, caspase-8, and JNK.
|
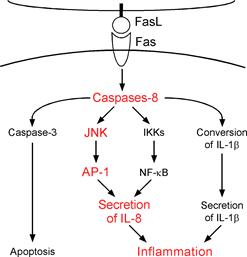
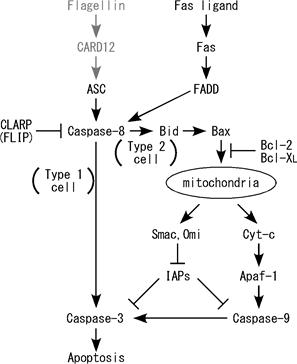
Figure. Fas ligand is well known death factor that
induces apoptosis in a caspase-8 dependent manner. We previously demonstrated
that stimulation by Fas ligand induces IL-1b
secretion in LPS-primed mouse macrophages and IL-8 secretion in human embryonic
kidney (HEK)-293 cells. Interestingly, it was found that both of these
inflammatory responses are caspase-8 dependent. In this study, we further
discovered that activation of Fas by Fas ligand induces AP-1 activation in a
caspase-8- and JNK-dependent manner. This AP-1 activation is required for the
Fas ligand-induced IL-8 production in HEK293 cells. |
|
Fas-Associated
Factor 1 is a negative regulator of PYRIN-containing Apaf-1-like protein 1
Takeshi
Kinoshita, Chiaki Kondoh, Mizuho Hasegawa, Ryu
Imamura, and
PYRIN-containing apoptotic protease-activating factor-1-like proteins (PYPAFs, also
called NALPs) participate in inflammatory signaling by regulating NF-kB activation and
cytokine processing, and have been implicated in autoimmune and inflammatory
disorders. However, the precise mechanisms that regulate the signal pathway
leading to NF-kB
activation are not completely understood. Here, we used yeast-two hybrid assays
to identify Fas associated factor 1 (FAF1) as a protein interacting with the
pyrin domains of several PYPAFs. In these assays, FAF1 interacted strongly with
PYPAF1, PYPAF3, and PYPAF7, moderately with PYPAF2 and PYNOD, but not at all
with the pyrin domains of pyrin or the adaptor molecule ASC. The interaction
between FAF1 and PYPAF1 in mammalian cells was confirmed by immunoprecipitation
assays, and the Fas-interacting domain of FAF1 was critical for this
interaction. When coexpressed in HEK293 cells, FAF1 interfere with NF-B
activation induced by PYPAF1 and ASC. A FAF1 mutant lacking the Fas-interacting
domain showed significantly reduced ability to inhibit NF-kB activation.
Furthermore, down-regulation of endogenous FAF1 protein augmented LPS-induced
IL-8 production, a biological marker for NF-kB activation, in
monocytic cells. Finally, the level of FAF1 expression in THP-1 cells increased
in response to NF-kB
stimulation. These findings suggest that FAF1 functions as a negative regulator
of an NF-kB
signal pathway that involves PYPAF1 and ASC.
|
|
|
|
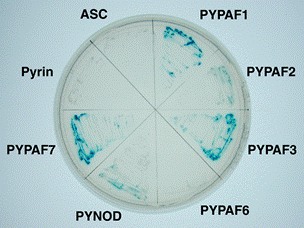
Figure 1. FAF1
interacts with the pyrin domains of PYPAF1, PYPAF2, PYPAF3, PYNOD, and
PYPAF7, but not with those of pyrin, ASC and PYPAF6 in Yeast-two hybrid
assays. |
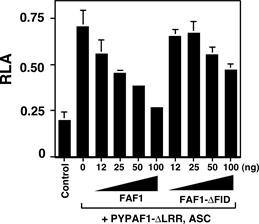
Figure 2. Cotransfection
of a constitutive active mutant of PYPAF1 (PYPAF1-DLRR) and ASC induces NF-kB activation as revealed by a luciferase
reporter assay in HEK293 cells. This NF-kB activation was inhibited by cotransfection of FAF1. |
Expression of NLRP7
(PYPAF3) protein in endometrial cancer tissues
Satoshi Ohno*, Takeshi Kinoshita*, Yumiko Ohno,
Toshinari Minamoto, Nobutaka Suzuki, Masaki Inoue and
Nucleotide-binding domain and leucine-rich
repeat-containing family, pyrin domain-containing 7 (NLRP7) (pyrin-containing
apoptotic protease activating factor-1-like protein 3; PYPAF3, NACHT domain-,
leucine-rich repeat, and pyrin domain-containing 7; NALP7) has been thought to
contribute to innate immunity and inflammation. Although expression of NLRP7 in
human seminoma tissues and several cancer cell lines has been demonstrated, the
pathophysiological and prognostic importance in cancer tissues has not been
defined. In this study, a series of 70 endometrial cancer cases that had
undergone curative resection was studied to determine the correlation between
NLRP7 expression and clinico-pathological
characteristics in human endometrial cancer tissue. Tissue specimens were
evaluated for NLRP7 by immunohistochemistry. NLRP7 expression was positive in
cancer cells in 7 cases (10%). There was a statistical relationship between the
depth of tumor invasion and NLRP7 expression (p=0.0326). NLRP7 expression
showed a trend for being associated with poor prognosis. Conclusion: Tumor-produced
NLRP7, associated with myometrial invasion, might
provide additional prognostic information in endometrial cancer patients.
|
|
|
|
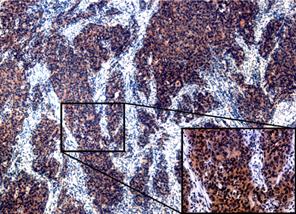
Figure 1.
Representative sections of endometrial cancer with immunohistochemical
staining of NLRP7. Strong cytoplasmic staining is observed in the invasion
front of the tumor (×40; inset, ×200). |
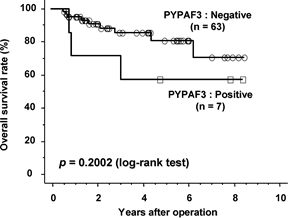
Figure 2. The
Kaplan-Meier survival curves of 70 patients with endometrial carcinoma in
relation to NLRP7 expression are shown. |
Mechanism of ASC-mediated apoptosis: Bid-dependent apoptosis in
type II cells
M. Hasegawa, K. Kawase, N. Inohara, R. Imamura, W-C. Yeh, T. Kinoshita, and T. Suda
ASC
is an adaptor molecule that mediates apoptotic and inflammatory signals, and
implicated in tumor suppression. However, the mechanism of ASC-mediated
apoptosis has not been well elucidated. Here, we investigated the molecular
mechanisms of ASC-mediated apoptosis in several cell lines using a CARD12-Nod2
chimeric protein that transduces the signal from muramyl dipeptide into
ASC-mediated apoptosis. Experiments using dominant-negative mutants,
small-interfering RNAs, and peptide inhibitors for caspases indicated that
caspase-8 was generally required for ASC-mediated apoptosis, while a
requirement for caspase-9 depended on the cell type. In addition, CLARP/FLIP (a
natural caspase-8 inhibitor) suppressed ASC-mediated apoptosis,
and Clarp-/- mouse embryonic
fibroblasts were highly sensitive to ASC-mediated apoptosis. Bax-deficient
HCT116 cells were resistant to ASC-mediated apoptosis as reported previously,
although we failed to observe colocalization of ASC and Bax in cells. Like
Fas-ligand-induced apoptosis, the ASC-mediated apoptosis was inhibited by Bcl-2
and/or Bcl-XL in type-II but not type-I cell lines. Bid was cleaved upon ASC
activation, and suppression of endogenous Bid expression using
small-interfering RNAs in type-II cells reduced the ASC-mediated apoptosis.
These results indicate that ASC, like death receptors, mediates two types of
apoptosis depending on the cell type, in a manner involving caspase-8.
|
|
c |
|
|
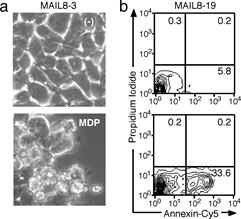
Figure. a, b) MDP induces apoptosis in MAIL8 cells expressing
CARD12-NOD2 chimera protein and ASC. c) ASC, like death receptors, mediates
two types of apoptosis depending on the cell type, in a manner involving
caspase-8 |
||
Publications
1.
Kinoshita,
T., Kondoh, C., Hasegawa, M., Imamura, R., and Suda,
T. (2006) Fas-associated Factor 1 is a negative regulator of PYRIN-containing
Apaf-1-like protein 1, Int. Immunol., 18:1701-1706.
2.
El Kasmi,
K.C., Holst J, Coffre, M., Mielke,
L., de Pauw, A., Lhocine,
N., Smith, A.M., Rutschman, R., Kaushal,
D., Shen, Y., Suda, T., Donnelly, R.P., Myers, M.G.
Jr., Alexander, W., Vignali, D.A., Watowich, S.S., Ernst, M., Hilton, D.J., Murray, P.J.
(2006) General nature of the STAT3-activated anti-inflammatory response. J. Immunol., 177:7880-7888.
3. Hasegawa, M., Kawase, K., Inohara, N.,
Imamura, R., Yeh, W-C.,Kinoshita,
T., and Suda, T. (2007) Mechanism of ASC-mediated apoptosis:
Bid-dependent apoptosis in type II cells, Oncogene, 26:1748-1756.
4.
Fujisawa, A., Kambe, N., Saito, M., Nishikomori, R., Tanizaki, H., Kanazawa,
N., Adachi, S., Heike, T., Sagara, J., Suda, T., Nakahata,
T., Miyachi, Y. (2007) Disease-associated mutations
in CIAS1 induce cathepsin B-dependent rapid cell death of human THP-1 monocytic
cells, Blood, 109:2903-2911.
5.
Umemura,
M., Yahagi, A., Hamada, S., Begum, M. D., Watanabe, H., Kawakami, K., Suda, T.,
Sudo, K., Nakae, S., Iwakura, Y., and Matsuzaki, G.
(2007) IL-17-mediated regulation of innate and acquired immune response against
pulmonary Mycobacterium bovis BCG infection, J.
Immunol., 178:3786-3796.
6. Matsumoto,
N., Imamura, R., Suda, T. (2007) Caspase-8- and JNK-dependent AP-1 activation
is required for Fas ligand-induced IL-8 production. FEBS J. 274:2376-2384
7. Ohno, S.*, Kinoshita, T.*, Ohno,
Y., Minamoto, T., Suzuki N., Inoue M., and Suda, T.
(2008) Expression of NLRP7 (PYPAF3, NALP7) Protein in
Endometrial Cancer Tissues. Anticancer Res. 28:2493-2497.
(* Both authors equally contributed to this work.)